exercise 04 :: Density of states and band structure calculations
DFT allows to calculate density of states (DOS) and energy band dispersion for crystalline solids. In the following exercise we show in detail the procedure for Al FCC crystal. We consider FCC Aluminium as introduced in exercise 02 and use converged self-consistent field (SCF) calculations parameters as the number of k-points and the ecutwfc parameter, see exercise 03. The pseudopotential file can be downloaded here: Al.pz-vbc.UPF.
TASK 01
Calculation flow for DOS is as follows:
-
perform
calculation = 'scf'(for converged set of parameters) usingpw.xprogram. Replace the?in the following input file with the converged values found in previous exercise.&control calculation = 'scf' restart_mode = 'from_scratch', pseudo_dir = './', outdir = './tmp', prefix = 'al' / &system ibrav = 2, celldm(1) = 7.652446, nat = 1, ntyp = 1, ecutwfc = ?, occupations = 'smearing', smearing = 'marzari-vanderbilt', degauss = 0.05 / &electrons diagonalization = 'david' mixing_beta = 0.7 conv_thr = 1.D-6 / ATOMIC_SPECIES Al 26.98 Al.pz-vbc.UPF ATOMIC_POSITIONS Al 0.00 0.00 0.00 K_POINTS (automatic) ? ? ? 0 0 0Save the file as
pw-scf.inand run the SCF calculations invokingpw.x < pw-scf.in > out.pw-scf. -
perform
calculation = 'nscf', i.e., non-selfconsistent calculations for larger number of k-point sampling of Brillouin zone. Remember to set also theecutwfcparameter in the following input:&control calculation = 'nscf' restart_mode = 'from_scratch', pseudo_dir = './', outdir = './tmp', prefix = 'al' / &system ibrav = 2, celldm(1) = 7.652446, nat = 1, ntyp = 1, ecutwfc = ?, occupations = 'smearing', smearing = 'marzari-vanderbilt', degauss = 0.001 / &electrons diagonalization = 'david' mixing_beta = 0.7 conv_thr = 1.D-6 / ATOMIC_SPECIES Al 26.98 Al.pz-vbc.UPF ATOMIC_POSITIONS Al 0.00 0.00 0.00 K_POINTS (automatic) 36 36 36 0 0 0Save the file as
pw-nscf.inand runpw.x < pw-nscf.in > out.pw-nscf -
save the following file as
dos.inand perform the DOS calculations usingdos.x < dos.in > out.dos&DOS prefix = 'al' outdir = './tmp' bz_sum = 'smearing' emin = -5.0 emax = 15.0 degauss = 0.01 deltaE = 0.01 fildos = 'K36_smear0.01_de0.01.dos' /
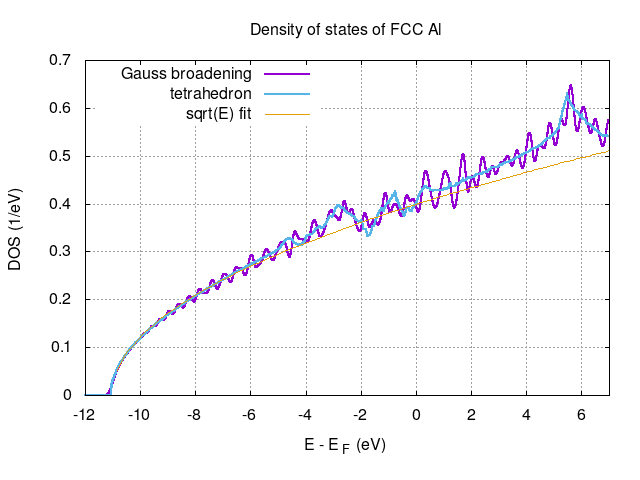
To improve on DOS calculation one might use larger number of k-points or consider tetrahedron method. For the tetrahedron method one needs to perform nscf calculations with occupations = 'tetrahedra' without setting smearing and degauss variables in the input for pw.x. Similarly for dos.in file one omits the degauss and sets bz_sum = 'tetrahedra'. Set also fildos = 'K36_tetra_de0.01.dos' Obtained results for both the integration schemes as well as the sqrt(E) fit are show in the following figure produced by the following gnuplot script
set term x11
set title "Density of states of FCC Al"
set ylabel "DOS (1/eV)"
set xlabel "E - E_F (eV)"
EF_nscf=7.6609 #read from out.pw-nscf
EF_tetra=7.6759 #read from out.pw-nscf_tetra
set key left
set grid
f(x) = a*sqrt(x-b)
a = 0.120294 # +/- 2.747e-05 (0.02283%)
b = -11.0382 # +/- 0.0006437 (0.005832%)
p [-12:7] \
'K36_smear0.01_de0.001.dos' u ($1-EF_nscf):2 t "Gauss broadening" w l lw 2,\
'K36_tetra_de0.01.dos' u ($1-EF_tetra):2 t"tetrahedron" w l lt 3 lw 2,\
f(x) t"sqrt(E) fit" w l lt 4
TASK 02
For band structure calculations one can proceed as follow:
-
perform
calculation = 'scf'(for converged set of parameters) usingpw.xprogram -
perform
calculation = 'bands'of eigenvalues along a k-path in the Brillouin zone usingpw.xprogram. The input might look like&control title = 'Al bands: L - G - K - W - X - U - L' calculation = 'bands' restart_mode = 'from_scratch', pseudo_dir = './', outdir = './tmp', prefix = 'al' / &system ibrav = 2, celldm(1) = 7.652446, nat = 1, ntyp = 1, ecutwfc = 50, occupations = 'smearing', smearing = 'marzari-vanderbilt', degauss = 0.001 / &electrons diagonalization = 'david' mixing_beta = 0.7 conv_thr = 1.D-6 / ATOMIC_SPECIES Al 26.98 Al.pz-vbc.UPF ATOMIC_POSITIONS Al 0.00 0.00 0.00 K_POINTS crystal_b 7 0.5 0.5 0.5 24 0.0 0.0 0.0 30 0.75 0.375 0.375 10 0.75 0.5 0.25 10 0.5 0.5 0.0 10 0.625 0.625 0.25 24 0.5 0.5 0.5 1To set coordinates of the high symmetry points for the k-path see video tutorial using XCrySDen or check the file or http://lamp.tu-graz.ac.at/~hadley/ss1/bzones/fcc.php
-
to collect the eigenvalues of the bands invoke
bands.x < bands.inwhere thebands.inis as follows:&bands prefix = 'al' outdir = './tmp' filband = 'bands.dat' lsym = .false. /The generated file
bands.dat.gnucan be plot using gnuplot script:set term x11 set title "Bandstructure of FCC Al" EF = 7.6599 #read from out.pw-scf set grid xtics set xtics ("L" 0, "{/Symbol G}" 0.82994101, "K" 1.89133024, "W" 2.24488363, "X" 2.73023897, "U" 3.09843702, "L" 3.74616479) set ylabel "E - E_F (eV)" p [:][-12:15] 'bands.dat.gnu' u 1:($2 - EF) t"" w l lw 2, 0 t"" lw 0.1 lt

Compare the calculated band structure with the empty lattice approximation.
Martin Gmitra :: martin.gmitra@upjs.sk
 This work is licensed under a Creative Commons Attribution 4.0 International License.
This work is licensed under a Creative Commons Attribution 4.0 International License.