exercise 06 :: Calculation of equilibrium structures of molecules
In order to investigate structure of molecules let us consider a simplest polyatomic system, a diatomic molecule, the nitrogen molecule N2. In order to study stability of the molecule we can introduce the binding energy in the form
which is the difference between U(d) the total potential energy of the molecule that depends on the N-N bond length d, and energy of the isolated nitrogen atom.
TASK 01
Calculate energy of the isolated N atom assuming unit cell of a cube shape of dimension 10 Anstrom for the three different exchange-correlation functionals - local density approximation (LDA), generalized gradient approximation (GGA) by Perdew-Burke-Ernzerhof (PBE), and revised Perdew-Burke-Ernzerhof GGA (PBESOL). For pseudopotentials use the projector augmented wave (PAW) pseudopotentials:
LDA (N.pz-n-kjpaw_psl.0.1.UPF)
PBE (N.pbe-n-kjpaw_psl.0.1.UPF)
PBESOL (N.pbesol-n-kjpaw_psl.0.1.UPF)
Enter the corresponding pseudopotential UPF files to the input file:
&control
calculation = 'scf'
restart_mode = 'from_scratch',
pseudo_dir = './',
outdir = './tmp',
prefix = 'N-atom'
tprnfor = .true.
tstress = .true.
/
&system
ibrav = 0,
nat = 1, ntyp = 1,
ecutwfc = 39, ecutrho = 265, occupations = 'smearing',
smearing = 'gauss', degauss = 0.02
/
&electrons
diagonalization = 'david'
mixing_beta = 0.7
conv_thr = 1.D-9
/
CELL_PARAMETERS Angstrom
10.0 0.0 0.0
0.0 10.0 0.0
0.0 0.0 10.0
ATOMIC_SPECIES
N 14.007 'enter here corresponding UPF file name'
ATOMIC_POSITIONS Angstrom
N 0.0 0.0 0.0
K_POINTS gamma
Compare your results to the following reference table:
| N atom | LDA | PBE | PBESOL |
|---|---|---|---|
| total energy | -26.80148 Ry | -27.64042 Ry | -27.20232 Ry |
TASK 02
Calculate binding energy of the N2 molecule as a function of the N-N separation using LDA exchange correlation functional. Prototypical input file for N-N distance of 1 Angstrom should like like:
&control
calculation = 'scf'
restart_mode = 'from_scratch',
pseudo_dir = './',
outdir = './tmp',
prefix = 'N2'
tprnfor = .true.
tstress = .true.
/
&system
ibrav = 0,
nat = 2, ntyp = 1,
ecutwfc = 39, ecutrho = 265, occupations = 'smearing',
smearing = 'gauss', degauss = 0.02
/
&electrons
diagonalization = 'david'
mixing_beta = 0.7
conv_thr = 1.D-9
/
CELL_PARAMETERS Angstrom
10.0 0.0 0.0
0.0 10.0 0.0
0.0 0.0 10.0
ATOMIC_SPECIES
N 14.007 N.pz-n-kjpaw_psl.0.1.UPF
ATOMIC_POSITIONS Angstrom
N 0.0 0.0 0.0
N 1.0 0.0 0.0
K_POINTS gamma
Hint: * For automated series of self-consistent field calculations for different N-N separation you can write your BASH script as in previous exercises or you can use PWTK scripting interface.
Calculations with PWTK
To install PWTK in your system download and unzip the package. Enter folder of the package and add the PWTK package folder to your working PATH using command export PATH=$PATH:$(pwd) (consider folder structure without blank spaces). Most probably you would need to install to your system, tcl components invoking: sudo apt-get install tcl tcllib*.
For PWTK package you need the following input pw_N2_scf.pwtk as follows:
CONTROL {
calculation = 'scf'
restart_mode = 'from_scratch',
pseudo_dir = './',
outdir = './tmp',
prefix = 'N2'
tprnfor = .true.
tstress = .true.
}
SYSTEM {
ibrav = 0,
nat = 2, ntyp = 1,
ecutwfc = 39, ecutrho = 265, occupations = 'smearing',
smearing = 'gauss', degauss = 0.02
}
ELECTRONS {
diagonalization = 'david'
mixing_beta = 0.7
conv_thr = 1.D-9
}
CELL_PARAMETERS Angstrom {
10.0 0.0 0.0
0.0 10.0 0.0
0.0 0.0 10.0
}
ATOMIC_SPECIES {
N 14.007 N.pz-n-kjpaw_psl.0.1.UPF
}
ATOMIC_POSITIONS Angstrom {
"N 0.0 0.0 0.0
N $d 0.0 0.0"
}
K_POINTS gamma {
}
And create PWTK control script scan_N2_bond_distance.pwtk:
# load the common definitions
source pw_N2_scf.pwtk
#-------------------------------------
# do series of calcs for separation d
#-------------------------------------
foreach d {0.50 0.60 0.70 0.80 0.90 1.00 1.10 1.20 1.40 1.60 1.80 2.00 2.25 2.50 2.75 3.00} {
# load the new N-N interatomic distance
ATOMIC_POSITIONS Angstrom " N $d 0.0 0.0 \n N 0.0 0.0 0.0"
# perform the calculation (io files: pw_N2_scf_d$d.scf.in|out)
runPW pw_N2_scf_LDA_d$d.scf
}
Once you have created the PWTK files you can run the series of calculations invoking command: pwtk scan_N2_bond_distance.pwtk. To get total energies for each separation grep for them using: grep "! " pw_N2_scf_LDA_d?.??.scf.out. Results can be plot using gnuplot as follows:
#set term png enahnced large font "Helvetica,12" #uncomment to save the png figure
E_N=-26.80148
Ry_to_eV=13.60569172
set title "binding energy of N_2 molecule"
set ylabel "binding energy [eV]"
set xlabel "N-N distance [Angstrom]"
set grid
p "< grep \"!\" pw_N2_scf_LDA_d?.??.scf.out | sed 's/pw_N2_scf_LDA_d//g' | sed 's/.scf.out:! total energy =//g'" using 1:(($2 - 2 * E_N)*Ry_to_eV) t"LDA" w lp pt 6

TASK 03
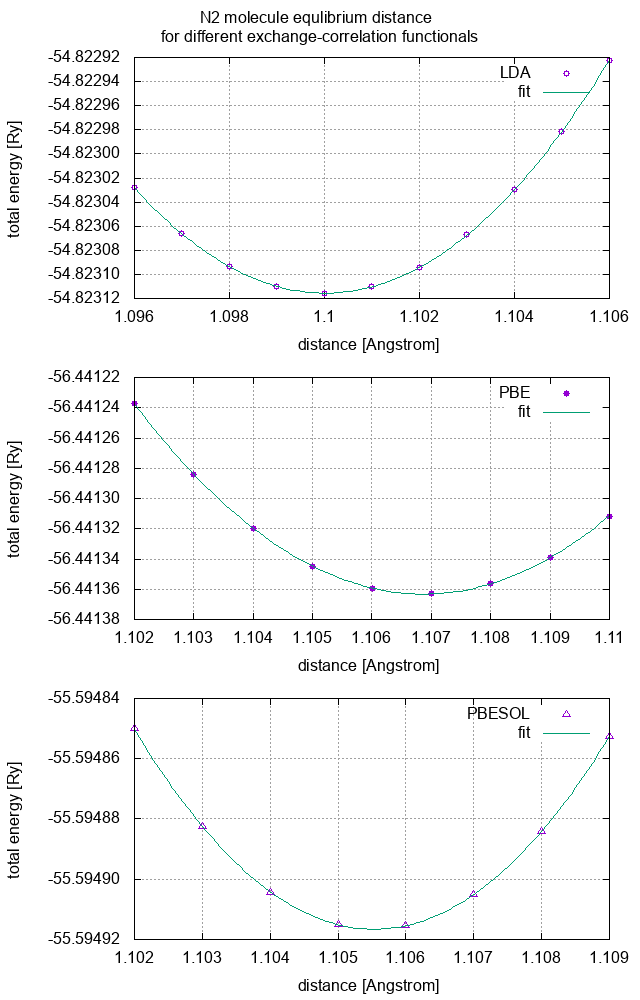
Find equilibrium bond distance of N2 molecule that corresponds to minimum of potential (total) energy for LDA, PBE and PBESOL exchange correlation functionals. As the total energy close to the equilibrium distance behaves parabolically you can fit the dependencies and the equilibrium distances obtain from the parabola minimum. For this purpose you can use gnuplot script
fLDA(x)=a_1*x*x + b_1*x + c_1
fit fLDA(x) "< grep \"!\" pw_N2_scf_LDA_d?.???.scf.out | sed 's/pw_N2_scf_LDA_d//g' | sed 's/.scf.out:! total energy =//g'" via a_1,b_1,c_1
p "< grep \"!\" pw_N2_scf_LDA_d?.???.scf.out | sed 's/pw_N2_scf_LDA_d//g' | sed 's/.scf.out:! total energy =//g'" u 1:2 t"LDA" w p pt 6, fLDA(x) t"fit" w l
Compare your results to reference data shown below and discuss dependence of the equilibrium distances on type of exchange-correlation functional used. Calculate binding energies of N2 molecule at equilibrium distances for LDA, PBE and PBESOL exchange correlation functionals. Discuss the results.
| N2 molecule | LDA | PBE | PBESOL |
|---|---|---|---|
| equlibrium distance [A] | 1.10001 | 1.10687 | 1.10553 |
| binding energy [eV] | ? | ? | ? |
The parabolic fits of the total energies as functions of N-N distance should look as follows:
Martin Gmitra :: martin.gmitra@upjs.sk
 This work is licensed under a Creative Commons Attribution 4.0 International License.
This work is licensed under a Creative Commons Attribution 4.0 International License.