exercise 05 :: Fermi surface, projected density of states, band structure and electron density of FCC Cu
The aim of the following exercise is demonstrate calculation of the Fermi surface, projected density of states (PDOS), band structure and electron density for Cu FCC crystal. For DFT calculations assume lattice constant of 3.597 Angstrom, for pseudopotential use Cu.pbe-dn-kjpaw_psl.1.0.0.UPF with the energy cutoff ecutoff = 45 and for the kinetic energy cutoff for charge density and potential ecutrho = 240.
TASK 01
Fermi surface calculation:
-
perform
calculation = 'scf'usingpw.xprogram. Set the?in the following input file. Be aware of units forcelldm(1)parameter.&control title = 'fcc Cu' calculation = 'scf' restart_mode = 'from_scratch', pseudo_dir = './', outdir = './tmp', prefix = 'cu' tprnfor = .true. tstress = .true. / &system ibrav = 2, celldm(1) = ?, nat = 1, ntyp = 1, ecutwfc = ?, ecutrho = ?, occupations = 'smearing', smearing = 'marzari-vanderbilt', degauss = 0.01 / &electrons startingpot = 'file' conv_thr = 1.D-7 diagonalization = 'david' mixing_beta = 0.7 / ATOMIC_SPECIES Cu 63.546 Cu.pbe-dn-kjpaw_psl.1.0.0.UPF ATOMIC_POSITIONS Cu 0.0 0.0 0.0 K_POINTS (automatic) 10 10 10 0 0 0
Find Fermi energy in the output file, e.g., grep Fermi out.pw-scf.
-
perform
calculation = 'nscf', i.e., non-selfconsistent calculations forK_POINTS (automatic) 30 30 30 0 0 0 -
Download c2x program and run it to extract the Fermi surface
./c2x --bxsf tmp/cu.xml copper.bxsf
To plot the Fermi surface use
xcrysden --bxsf copper.bxsf
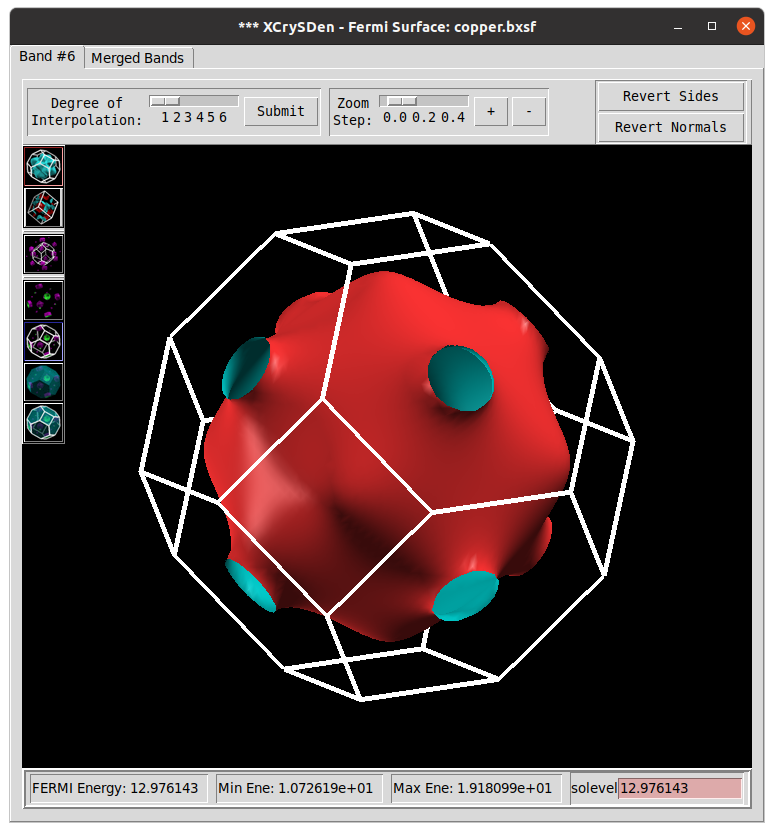
TASK 02
PDOS on particular atomic orbital can be written as follows
where the Bloch wave function is projected to an atomic orbital with a specific orbital momentum. For DOS and PDOS calculation we use tetrahedron method setting occupations = 'tetrahedra' (removing smearing and degauss) and perform non-selfconsistent calculations setting calculation = 'nscf' to obtain electronic density for further post-processing.
-
For total DOS calculation use the following input for
dos.xprogram:&DOS prefix = 'cu' outdir = './tmp' bz_sum = 'tetrahedra' emin = 0.0 emax = 25.0 deltaE = 0.1 fildos = 'K30_tetra_de0.1.dos' / -
For PDOS calculations we will use
projwfc.xprogram (see documentation for input parameters ). The input fileprojwfc.inis as follows:&projwfc prefix = 'cu', outdir = './tmp' filproj = 'pdos.proj' /To run the PDOS calculations invoke:
projwfc.x < projwfc.in > out.projwfc_pdos. -
To plot the DOS and PDOS you can use the following gnuplot script:
set term x11 set title "Density of states of FCC Al" set ylabel "DOS (1/eV)" set xlabel "E - E_F (eV)" set style fill noborder set style fill transparent solid 0.35 noborder EF=12.8277 set key right set grid p [-10:10] \ 'K30_tetra_de0.1.dos' u ($1-EF):2 t"total" w l lt 1 lw 2,\ 'cu.pdos_atm#1(Cu)_wfc#3(d)' u ($1-EF):2 t"d-orbital" w filledcurves lc rgb "forest-green",\ 'cu.pdos_atm#1(Cu)_wfc#2(p)' u ($1-EF):2 t"p-orbital" w l lt 3 lw 2,\ 'cu.pdos_atm#1(Cu)_wfc#1(s)' u ($1-EF):2 t"s-orbital" w l lt 2 lw 2

TASK 03
Band structure calculations for the FCC Cu is similar to previous exercise 04 resulting in the following plot:
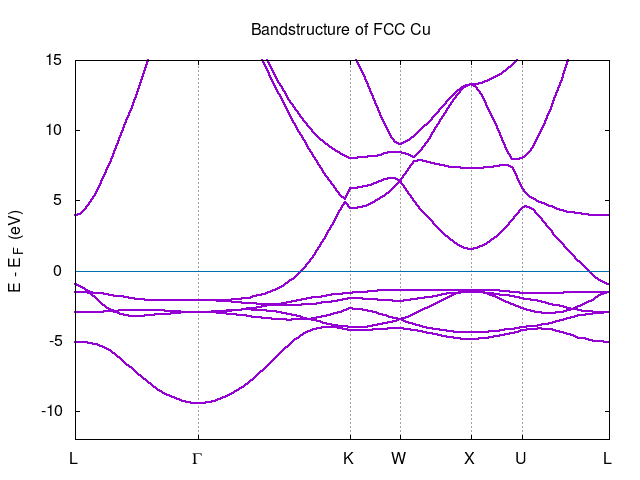
TASK 04
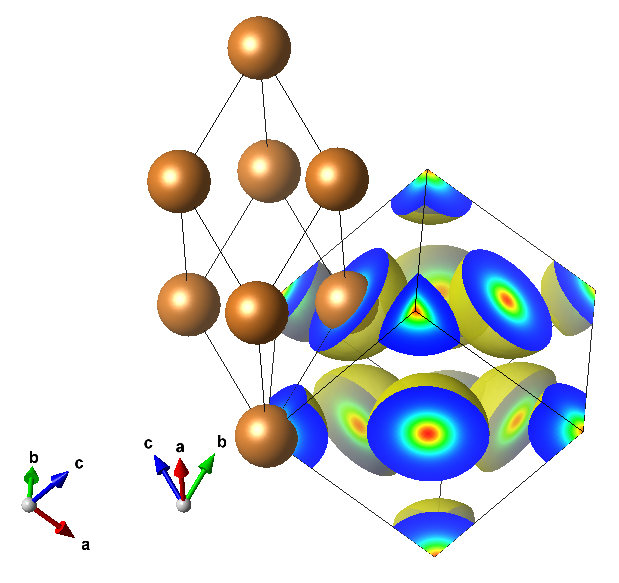
Electron density plot can be performed using pp.x < edp.in where the input file is as follows:
&inputpp
prefix = 'cu'
outdir='./tmp/',
filplot = 'tmp.pp'
plot_num = 0,
/
&plot
nfile=1
filepp(1)='tmp.pp', weight(1)=1.0
iflag=3
output_format=3
e1(1)=1.0, e1(2)=0.0, e1(3)=0.0
e2(1)=0.0, e2(2)=1.0, e2(3)=0.0
e3(1)=0.0, e3(2)=0.0, e3(3)=1.0
x0(1)=0.0, x0(2)=0.0, x0(3)=0.0
nx=50, ny=50, nz=50
fileout='edp_3d.xsf'
/
The resulting electron density plot stored in the edp_3d.xsf can be plot by XCrysDen or VESTA program:
Martin Gmitra :: martin.gmitra@upjs.sk
 This work is licensed under a Creative Commons Attribution 4.0 International License.
This work is licensed under a Creative Commons Attribution 4.0 International License.