exercise 08 :: Calculation of magnetic properties of bcc Fe and FeO
The present exercise aims to demonstrate the self-consistent field calculations for cubic bulk bcc Fe and FeO crystals. We demonstrate calculation od spin polarized band structure and density of states, as well as inclusion of Hubbard U correction to on-site energy important for treating correlation effects in d-shell for FeO.
TASK 01
Consider bcc Fe with lattice constant of 2.856 Angstrom and perform self-consistent field calculations for non-magnetic case. The input file could look like
&control
calculation = 'scf'
restart_mode = 'from_scratch'
prefix = 'Fe_NM'
pseudo_dir = './'
outdir = './tmp'
/
&system
ibrav = 3, celldm(1) = 5.3970579, nat = 1, ntyp = 1
ecutwfc = 64, ecutrho = 782
occupations = 'smearing', smearing = 'marzari-vanderbilt', degauss = 0.05
/
&electrons
diagonalization = 'david'
conv_thr = 1.0e-8
mixing_beta = 0.6
/
ATOMIC_SPECIES
Fe 55.847 Fe.pbe-spn-kjpaw_psl.0.2.1.UPF
ATOMIC_POSITIONS alat
Fe 0.0 0.0 0.0
K_POINTS automatic
8 8 8 0 0 0
To perform self-consistent field calculations of ferromagnetic phase one sets up the following variables to &system block
nspin = 2
starting_magnetization(1) = 1.5
in which you instruct the code to perform spin-polarized calculations with starting magnetization on first atom in the ATOMIC_SPECIES list.
Remember also to change prefix in case you would like to use converged electronic density of ferromagnetic case for further postprocessing.
Consider now antiferromagnetic order of magnetic moments on the neighboring Fe atoms and perform self-consistent field calculation of the total electronic energy. Hint: for the calculation you need to modify the structure considering a cubic primitive cell and two non equivalent Fe atoms and setting the starting magnetization in opposite directions.
Compare your results for the total electronic energies with the following table and discuss the magnetic ground state.
| bcc Fe | non-magnetic | ferromagnetic | antiferromagnetic |
|---|---|---|---|
| total energy per atom (Ry) | -329.2312 | -329.2596 | -329.2343 |
Consult results for total magnetization and absolute magnetization values for the magnetic cases. The ferromagnetic phase is the ground state with magnetic moment per Fe atom of 2.14 with good agreement with experimental value 2.22
.
Exchange integral from total energy
The exchange integral can be determined by mapping the DFT results to the Heisenberg spin model Hamiltonian meaning we require that the total energies are equal. The Hamiltonian in this case reads
where is the exchange integral and
is the spin angular momentum operator at site i.
Assuming the nearest neighbors, in our case each Fe atom has the 8 neighbours, and the energy of the ferromagnetic state is thenand for antiferromagnetic state
. The exchange integral is equal
. The Curie temperature within the mean field approximation is
which for the bcc Fe leads to overestimating experimental value.
TASK 02
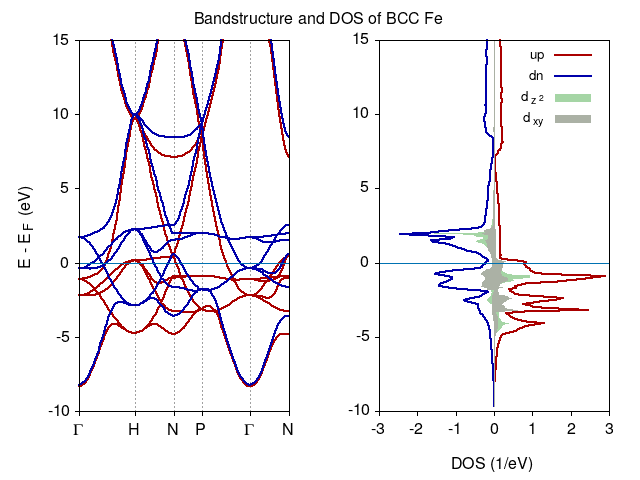
Calculate density of states (DOS), projected density of states (PDOS) considering Fe d-orbitals and band structure for ferromagnetic ground state of bcc Fe.
To calculate DOS and PDOS one needs to perform calculation = 'nscf' with larger k-point Brillouin zone sampling. Consider k-point sampling with 12x12x12 and tetrahedron method setting occupations = 'tetrahedra' similar inputs we prepared in exercise 05.
For total DOS calculations using dos.x we consider the following input
&DOS
prefix = 'Fe_FM'
outdir = './tmp'
bz_sum = 'tetrahedra'
emin = 8.0
emax = 33.0
deltaE = 0.1
fildos = 'tetra_de0.1.dos'
/
and for PDOS using projwfc.x program
&projwfc
prefix = 'Fe_FM',
outdir = './tmp'
filproj = 'pdos.proj'
emin = 8
emax = 33
/
To calculate band structure calculation = 'bands' along the high symmetry lines connecting (for the point coordinates see Sec. 1.3 in W. Setyawan and S. Curtarolo)
use:
K_POINTS crystal_b
6
0.0 0.0 0.0 34 G
0.5 0.5 0.5 28 H
0.5 0.0 0.0 17 N
0.25 -0.25 0.25 17 P
0.0 0.0 0.0 20 G
0.5 0.0 0.0 1 N
The bands.x program should be run afterwards separately for spin up and spin down specifying spin_component = 1 and spin_component = 2 in the input:
&bands
prefix = 'Fe_FM'
outdir = './tmp'
filband = 'bands_up.dat'
spin_component = ?
lsym = .false.
/
To plot the results you can use the following gnuplot script
set term x11
set multiplot layout 1,2 title "Bandstructure and DOS of BCC Fe"
#BANDS subfigure
EF=17.6895645097 #taken from tetra_de0.1.dos file
set grid xtics
set xtics ("{/Symbol G}" 0, "H" 1.0, "N" 1.7071067812, "P" 2.2071067812, "{/Symbol G}" 3.0731321850, "N" 3.7802389662) out nomirror offset 0,0.25
set ytics out nomirror offset 0.75,0
set xlabel " "
set ylabel "E - E_F (eV)" offset 1,0
set linestyle 1 lc rgb "#aa0000" lt 1 lw 2
set linestyle 2 lc rgb "#0000aa" lt 1 lw 2
p [:][-10:15] \
'bands_up.dat.gnu' u 1:($2-EF) t"" w l ls 1, 'bands_dn.dat.gnu' u 1:($2-EF) t"" w l ls 2 ,\
0 t"" lw 0.1 lt 6
#DOS subfigure
set xtics auto
unset ylabel
set xlabel "DOS (1/eV)"
set style fill noborder
set style fill transparent solid 0.35 noborder
set key right spacing 1.25 font "Helvetica,10"
unset grid
set linestyle 3 lc rgb "#008800" lt 1 lw 2
set linestyle 4 lc rgb "#112200" lt 1 lw 2
p [-3:3][-10:15] \
'tetra_de0.1.dos' u ($2):($1-EF) t"up" w l ls 1, '' u (-$3):($1-EF) t"dn" w l ls 2,\
'Fe_FM.pdos_atm#1(Fe)_wfc#5(d)' u 4:($1-EF) t"d_{z^2}" w filledcurves ls 3, '' u (-$5):($1-EF) t"" w filledcurves ls 3,\
'' u 12:($1-EF) t"d_{xy}" w filledcurves ls 4, '' u (-$13):($1-EF) t"" w filledcurves ls 4,\
0 t"" lw 0.1 lt 6
unset multiplot
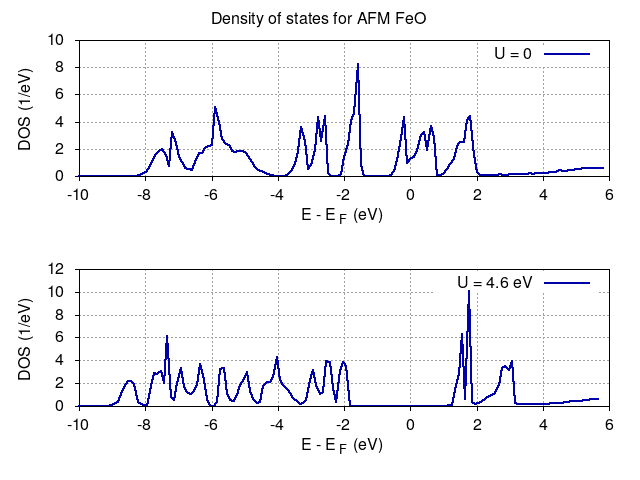
TASK 03
Calculate DOS for antiferromagnetic rock-salt cubic crystal FeO and compare results with on-site Hubbard potential U=4.6 eV for d electrons of Fe.
The input for the self-consistent field calculations reads as follows:
&control
calculation='scf'
restart_mode='from_scratch',
prefix='FeO'
pseudo_dir = './'
outdir='./tmp/'
verbosity='high'
/
&system
ibrav = 0,
celldm(1) = 8.19,
nat = 4, ntyp = 3,
ecutwfc = 40, ecutrho = 460,
occupations = 'smearing', smearing = 'mv', degauss = 0.02,
nspin = 2,
starting_magnetization(1) = 0.5,
starting_magnetization(2) = -0.5
lda_plus_u = .true.,
lda_plus_u_kind = 0,
U_projection_type = 'atomic',
Hubbard_U(1) = 4.6
Hubbard_U(2) = 4.6
starting_ns_eigenvalue(1,1,1) = 1.2d0
starting_ns_eigenvalue(2,1,1) = 1.2d0
starting_ns_eigenvalue(3,1,1) = 0.d0
starting_ns_eigenvalue(4,1,1) = 1.2d0
starting_ns_eigenvalue(5,1,1) = 0.d0
starting_ns_eigenvalue(1,2,2) = 1.2d0
starting_ns_eigenvalue(2,2,2) = 1.2d0
starting_ns_eigenvalue(3,2,2) = 0.d0
starting_ns_eigenvalue(4,2,2) = 1.2d0
starting_ns_eigenvalue(5,2,2) = 0.d0
nbnd = 50
/
&electrons
conv_thr = 1.d-9
mixing_beta = 0.3
mixing_fixed_ns = 8
/
ATOMIC_SPECIES
Fe1 55.845 Fe.pbe-spn-kjpaw_psl.0.2.1.UPF
Fe2 55.845 Fe.pbe-spn-kjpaw_psl.0.2.1.UPF
O 16.0 O.pbe-n-kjpaw_psl.1.0.0.UPF
CELL_PARAMETERS {alat}
0.50 0.50 1.00
0.50 1.00 0.50
1.00 0.50 0.50
ATOMIC_POSITIONS {crystal}
Fe1 0.00 0.00 0.00
Fe2 0.50 0.50 0.50
O 0.25 0.25 0.25
O 0.75 0.75 0.75
K_POINTS {automatic}
3 3 3 0 0 0
Density of states for U=0 and U=4.6 eV are shown below demonstrates spectral gap opening and change from metallic to insulating phase in accordance with experiment.
Martin Gmitra :: martin.gmitra@upjs.sk
 This work is licensed under a Creative Commons Attribution 4.0 International License.
This work is licensed under a Creative Commons Attribution 4.0 International License.